شبیه سازی دینامیک مولکولی
شبیه سازی ديناميك مولكولي در ابتدا توسط آلدر و وينرايت در اواخر دههي 1950 به منظور مطالعه ي برهمکنشهاي مدلي از کرات سخت معرفي شد . سپس در سال 1959، این روش برای اولین بار به منظور شبیه سازی یک ماده واقعی، اثر تشعشعات مس بلوری بر روی آثار تاریخی، توسط وینیارد مورد استفاده قرار گرفت. پيشرفت عمدهِ ي ديناميک مولکولي با کارهاي رحمان، که از وی به عنوان پدر دینامیک ملکولی یاد میشود، با شبيه سازي آرگون مايع و استفاده از پتانسيل لنارد-جونز صورت گرفت. دینامیک مولکولی روشی مناسب برای محاسبه خواص تعادلی و انتقالی سامانه ها می باشد. هدف اصلي در شبیه سازی ديناميک مولکولي بررسي حرکت همه ي ذرات موجود در سامانه مدل، تحت نيروهاي بين ذرات است که از طريق حل پی در پی معادلات حرکت نيوتني در n گام زماني متوالي حاصل ميشود و در نهايت n تصوير لحظه اي از سامانه مورد نظر را به وجود مي آورد. روشهای ديناميک مولکولي شامل دو شکل عمومی است که برای سامانه هاي تعادلی و غيرتعادلي استفاده میشوند. اما باید به این نکته توجه داشت که روش شبیه سازی دینامیک مولکولی همانند سایر روشها با خطا همراه می باشد. برای کاهش میزان خطاهای آماری می توان زمان شبیه سازی یا تعداد دفعات اندازه گیری را افزایش داد تا مقدار متوسط بدست آمده دقت بیشتری داشته باشد. در نهایت از مقایسه بین نتایج روش شبیه سازی دینامیک مولکولی و نتایج تجربی، می توان به میزان تطابق و صحت مدل (پتاسیل) مورد استفاده در شبیه سازی با واقعیت پی برد. اگر مدل مناسب و نزدیک به واقعیت انتخاب شده باشد، می تواند به تفسیرو توصیف نتایج و همچنین مشاهدات تجربی سامانه، کمک شایانی نماید .
برخی از اصول مورد استفاده در شبیه سازی دینامیک مولکولی
مراحل کلی انجام یک شبیه سازی دینامیک مولکولی در شکل زیر آورده شده است. از آنجا که در فصول بعد به تکرار با بعضی از اصطلاحات شبیه سازی سروکار داریم در ادامه به شرح مختصری در مورد آنها خواهیم پرداخت.
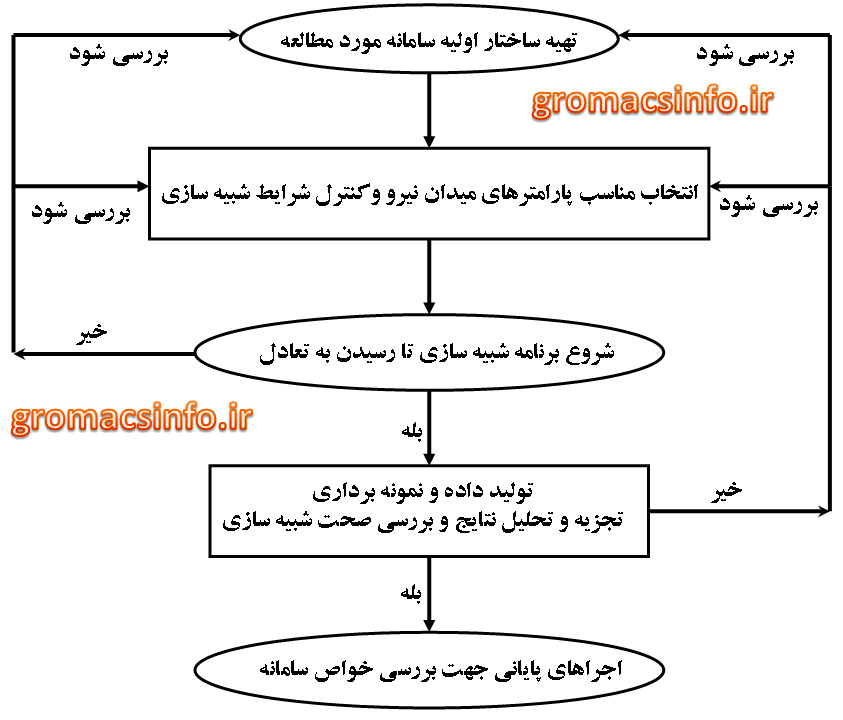
نمای کلی از مراحل شبیه سازی دینامیک مولکولی
الف- تهیه ساختار اولیه
همان گونه که در شکل فوق آورده شده است، مرحله اول در هر شبیه سازی، تهیه ساختار اولیه سامانه می باشد. این مرحله نقش تعیین کنندهای در زمان رسیدن به تعادل و صحت شبیه سازی دارد. پس این مرحله باید به دقت انجام شود. تعیین اتفاقی مکانهای اولیه ذرات، سبب همپوشانی اتمهای مجاور می شود که این موضوع باعث شکست محاسبات عددی در الگوریتم های دینامیک مولکولی می گردد. بنابراین از مقادیر تصادفی برای تهیه ساختار اولیه نمی توان استفاده کرد. اگر سامانه مورد مطالعه جامد باشد الزاماً باید آرایش اولیه ذرات با حالت واقعی سامانه انطباق داشته باشد که به راحتی می توان با استفاده از داده بلور نگاری، ساختار اولیه سامانه مورد نظر را تهیه کرد.اگر ساختار جامد توسط بلورنگاري پرتو ايکس مشخص شده باشد براي ساختن سلول واحد مي توان از نرم افزارهايي مثل مرکوری کمک گرفت. براي تهیه ساختار اوليه سامانه هاي همگن در حالت مايع يا گاز، وجود اطلاعات تجربي ساختار بلوري ماده ضرورت ندارد. در اين حالت اغلب از يک ساختار شبکه اي استاندارد (مثل FCC ) به عنوان ساختار اوليه استفاده مي شود. براي تعيين موقعيت اتم هاي سازنده مولکول نسبت به هم مي توان از ساختار بهينه شده مولکول توسط نرم افزار گوسين با يک سطح محاسباتي بالا استفاده نمود. روش ديگر براي توليد ساختار اوليه سامانه هاي مايع يا گازي، استفاده از ساختار بلوري جامد مي باشد. در اين روش با بالا بردن دما، سامانه جامد ذوب شده و آرايش تصادفي حاصل مي شود که از آن براي شروع شبيه سازي حالت مايع يا گازي استفاده مي شود. گاهي اوقات نيز براي شروع يک شبيه سازي مي توان از پيکربندي نهايي شبيه سازي يک سامانه در حالت تعادل شبيه به سامانه ي مورد نظر استفاده کرد. البته این نکته شایان ذکر است که برای پروتئینها می توان از پایگاه داده ای RCSB فایل ساختاری پروتئین مورد نظر رو به فرمت pdb دانلود کرد که این فایل از روشهای کریستالوگرافی ایکس ری یا روزنانس مغناطیسی هسته بدست آمده است. برای آشنایی بیشتر با این فایل فرمت روی اینجاکلیک کنید.
 استفاده از مطالب فوق با ذكر منبع بلامانع است!
استفاده از مطالب فوق با ذكر منبع بلامانع است!